
Reform of EU Medicines Legislation: A Blessing or a Missed Opportunity?
Authored by:

Meryem Uzunay
Associate Consultant

Erik Doevendans
Technical Head (NL) & Principal Consultant
Introduction
Ideally, all patients in the European Union should have equal access to safe, efficacious, and high-quality medicines. However, reality is more erratic, as many European citizens are all too familiar with the frustration of limited access to medicines. Patients, on average, wait up to 4 months to locate a given medicinal product or therapeutic in their nearest pharmacy, however, one could also wait for more than 2 years for the same product. Additionally, there is the growing concern regarding possible shortages of medicinal products, including essential products such as antibiotics and painkillers.
With the intention to solve access and supply issues, but also to stimulate innovation focussed on unmet medical needs, and to harmonise the EU internal market for the supervision and control of medicinal products; the European Commission (the Commission) aims to modernise the pharmaceutical sector with a patient-centred approach that also fully supports an innovative and competitive industry. Furthermore, regulatory process will be streamlined, data protection duration will be more incentive-driven, and systems and procedures will be implemented to foster innovation.
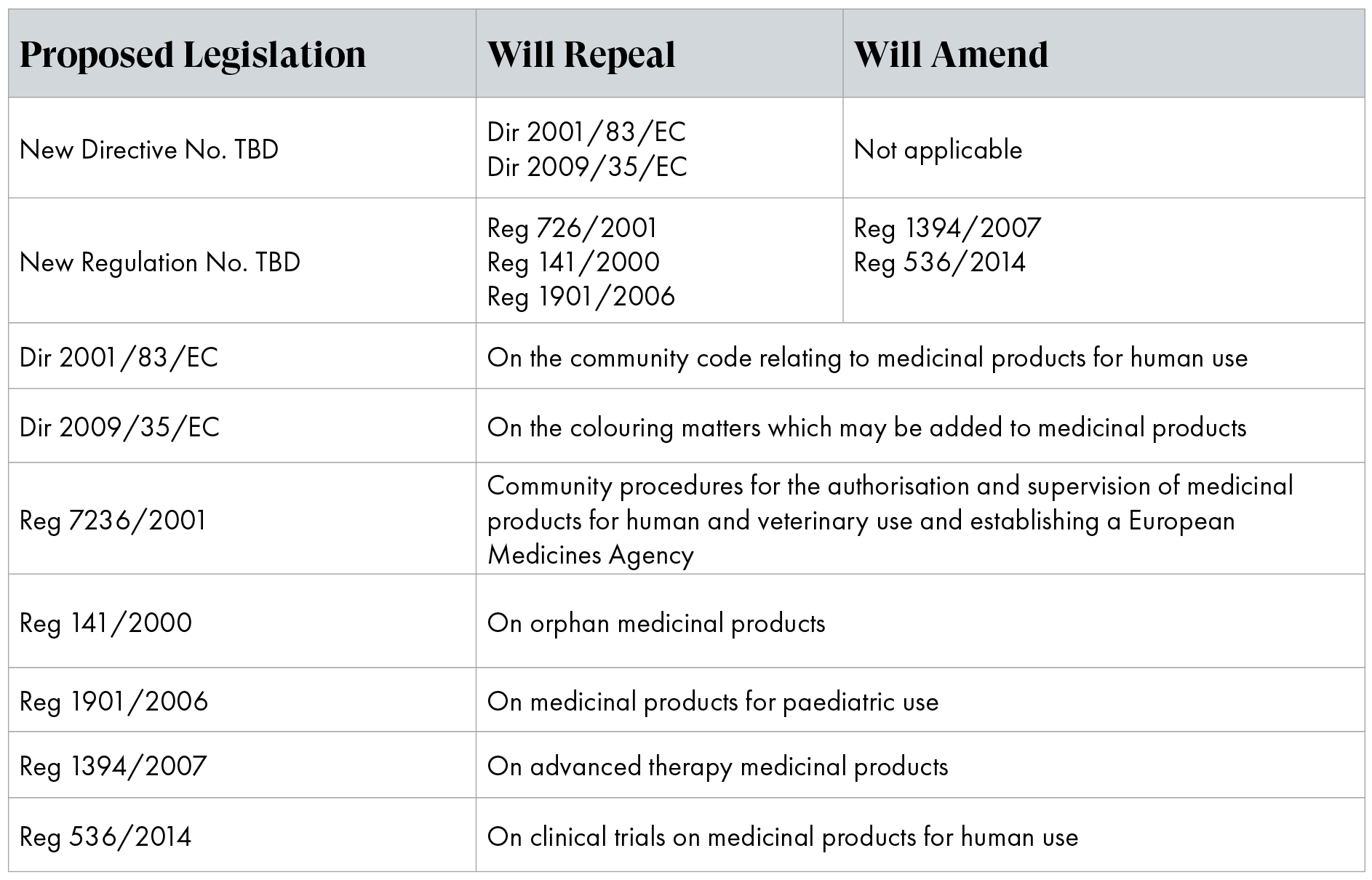
To facilitate the necessary changes, the Commission adopted and disclosed a proposal for a new Directive and a new Regulation. This proposal revises and replaces the existing general pharmaceutical legislation, including the legislation on rare diseases and children (Table 1).
Table 1: Legislation as proposed to be amended/repealed
Access to Medicines
With the reform, it is proposed that all marketing authorisation holders (MAHs) should have medicine shortage prevention plans in place, with the European Medicines Agency (EMA) providing guidance on approaches to streamline the implementation of those plans. In addition, Member States’ competent authorities should be empowered to monitor shortages based on notifications of MAHs.
Additionally, a program for the development of antibiotics will be initiated to overcome growing antimicrobial resistance and immunity. To qualify, the new antibiotic must be considered a “priority antimicrobial” suggesting it is required to show significant clinical benefit in overcoming antimicrobial resistance, and must either belong to a new class, have a different mechanism of action compared to existing therapies, and/or tackle infections that are difficult to treat.
For developers of “game-changing” novel antimicrobials, the reform introduces a voucher system that will provide transferable data exclusivity and offer an additional year of data protection to the developer, exemplifying that the proposed regulatory data protection duration will be more incentive-driven.
Regulatory Data Protection & Market Exclusivity
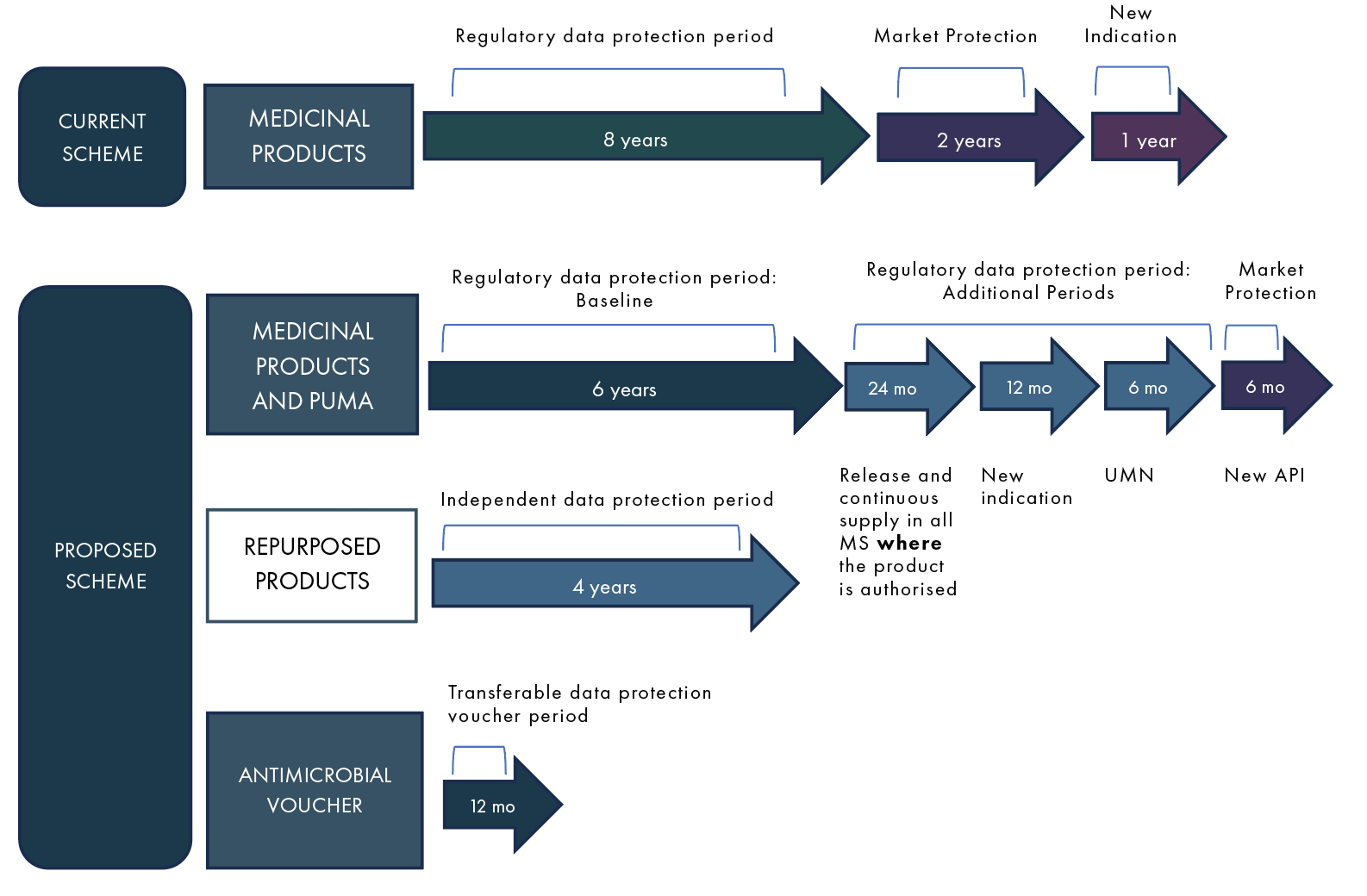
In general, the proposal introduces a reduction of the baseline data protection period, while extending the number of possibilities of prolongation of this period. This would lead to a reduction of the baseline data protection by 2 years resulting in 6 years data protection compared to 8 years today. Consequently, innovation and/or providing access to all patients in the Union will be rewarded (Table 2 and Figure 1).
Table 2: Proposed data protection duration

Furthermore, the proposed Regulation tends to soften the orphan’s market protection. The duration of market exclusivity would be 10 years for orphan medicines addressing a high unmet medical need, 5 years for orphan drugs that were registered based on bibliographic data, and 9 years for all others. A prolongation of exclusivity of 12 months could be obtained if the medicines were released and contiguously supplied within 2 years after approval, thereby covering the needs of patients, or if a new therapeutic indication was developed (at least two years before the initial exclusivity period ended). New principles will be in place for generic and biosimilar development of orphan drugs in addition to transitional provisions regarding orphan designations.
Repurposing medicinal products that are already authorised, for another indication, will be given a data protection of 4 years. It may be granted only once for any given product.
Figure 1: Current and proposed scheme of EU Registration Data Protection
MS= Member State(s); UMN= Unmet Medical Need;
PUMA= paediatric-use marketing authorisations; API = Active pharmaceutical ingredient
Streamlining Regulatory Procedures
With the reform, the EMA aims to provide guidance to MAHs on approaches to streamline the implementation of those plans. This includes the proposal for a reduction of review time from 210 days to 180 days for the assessment of marketing authorisation applications, and a further 46 days for authorisation by the Commission; the ‘perfect dossier’ will be promoted by the Agency, also through early agency interactions. Furthermore, the proposal aims at optimising the regulatory support (e.g., scientific advice) to Small and Medium-sized Enterprises (SMEs) and non-commercial organisations, resulting in additional reductions of administrative costs for these parties.
Sandbox
A regulatory sandbox is a time-limited regulatory framework, which enables the testing of innovative technologies, products, approaches, or services in a real-world environment under regulatory supervision to facilitate the development and authorisation of innovative products. The legal basis of a regulatory sandbox is a so-called “experimentation clause” allowing exemptions from existing requirements to try out novel regulatory solutions. With the sandbox, EMA and the Commission offer an environment to test adaptability of the pharmaceutical frameworks for new cutting-edge product developments.
Concerns & Outlook
Although the issued draft legislation suggests changes to the regulatory framework in an attempt to foster innovation by providing a more flexible environment, Scendea feels that there may also be some missed opportunities, such as filling the void between the industrial scale of manufacture and magistral pharmacy preparations. The reason for this category falling in between is due to their personalised nature, or because the disease indication is very rare (ultra-orphan disease). In these cases, a more pragmatic assessment of quality, safety and efficacy would have been prudent. Furthermore, considering the current experience with biotechnology products and following FDA’s approach with these products (now under CDER) it might have been an improvement if biotechnology products were also allowed to obtain marketing authorisation in individual EU Member States instead of the current mandatory Centralised Procedure.
Also, stakeholders’ appreciation of the proposal is not overly positive. According to the Proposal’s Impact Assessment Report [SWD(2023) 192 final - a Commission Staff Working Report], SMEs may experience difficulties in adjusting to a modulation of incentives linked to market launch due to their lack of capacity to serve all Member States in a timely manner.
Additional concerns were expressed about the Commission’s proposal to reduce the Regulatory Data Protection period from 8 to 6 years, given that the 8-year protection has been crucial for driving the development of innovative therapies and potentially could discourage companies from investing in R&D for the development of new treatments in Europe. Additional concerns from the European Federation of Pharmaceutical Industries and Associations (EFPIA) and the Hellenic Association of Pharmaceutical Companies (SFEE) include the impact of the policies set out across these proposals, on European competitiveness against mainly the USA and China, innovation, and patient access. Other stakeholders have a negative view on the introduction of vouchers to encourage the development of new antimicrobials, as they believe that this could prolong pharmaceutical monopolies, undermine generic competition, and severely slow down patient access to innovative and affordable therapeutics. The Commission’s Proposal is currently being discussed in the EU Parliament where lawmakers have until 14 November 2023 to present the amendments in the health committee aiming to reach a common position and move the discussion to the plenary. Considering the emerging controversy between Member States and the impact this legislation will have on important stakeholders, it remains to be seen whether or when its adoption will be achieved.
Conclusion
The proposal for a new Directive and a new Regulation aims to provide a solution to access and supply issues, while simultaneously driving innovation and harmonisation of the EU internal market for medicinal products.
While the proposal enables improved access to medicines by providing guidance on approaches to have medicine shortage prevention plans in place for MAHs, it drives this enhanced access further by introducing programs for the development of antibiotics with the aim to overcome growing antimicrobial resistance.
Furthermore, this patient-centred and innovation-driven approach is further emphasised through the streamlining of regulatory procedures by reducing the review time for the assessment and authorisation of marketing authorisation applications. Optimisations to the regulatory support, such as scientific advice, is also associated with additional cost reductions.
Furthermore, the revised regulatory data protection and market exclusivity leads to a reduced baseline data protection period by 2 years and prolongs market exclusivity by 12 months, which consequently results in rewarded innovation and access to all patients in the Union.
Finally, the legal basis of a regulatory sandbox allows exemptions from existing requirements to try out novel regulatory solutions. Therefore, this offers an environment to test adaptability of the pharmaceutical frameworks for new cutting-edge product developments.
In conclusion, the proposal focuses on a patient-centred approach, which also supports a competitive industry, the proposal streamlines regulatory processes, makes data protection duration incentive-driven, and implements systems and procedures to foster innovation.